Introducción
El nefroma mesoblástico congénito (NMC) es un tumor sólido con bajo potencial de malignidad que representa el 3-4% de los tumores renales pediátricos1,2. Su diagnóstico ocurre principalmente en neonatos y lactantes menores de 6 meses, aunque es raro, con una incidencia de 8 por millón de recién nacidos3. Se distinguen tres subtipos histológicos: clásico, celular y mixto; el clásico es de mejor pronóstico4.
La mayoría de los diagnósticos se realizan antes del primer año de vida, en general por una masa abdominal palpable. En algunos casos, los padres reportan hematuria o se detecta la masa en ecografías prenatales, junto con posibles manifestaciones de metástasis, como hidrocefalia. Estos tumores pueden asociarse con síndromes paraneoplásicos, expresados clínicamente como hipertensión arterial e hipercalcemia, y generar inestabilidad cardiovascular5.
Presentamos dos casos de NMC tratados con nefrectomía radical en el Instituto Nacional de Salud del Niño de Lima, Perú, con el objetivo de discutir la presentación clínica, el tratamiento quirúrgico y el seguimiento necesario para estos casos infrecuentes.
Casos clínicos
Paciente 1
Lactante de sexo femenino, menor de 2 meses, procedente de Lima. Madre de 22 años sin antecedentes relevantes ni complicaciones gestacionales. Nacida de parto eutócico con Apgar 9/10, y peso y talla adecuados. Acudió a emergencia del hospital con 2 horas de enfermedad por vómito lácteo y rasgos de sangre en la parte anterior del pañal. En la exploración física se evidenció palidez y una masa indurada, no dolorosa, en el flanco izquierdo que cruzaba la línea media (Fig. 1A), y peloteo renal. Laboratorio: leucocitos 11.630 cel/μl, linfocitos 66%, hemoglobina 12,8 g/dl, urea 12 mg/dl y creatinina 0,36 mg/dl. Uroanálisis: leucocitos 5-10 por campo, hematíes 3-5 por campo, pH 5, eritrocitos 3+ y esterasa leucocitaria 2+. Urocultivo: negativo para gérmenes.
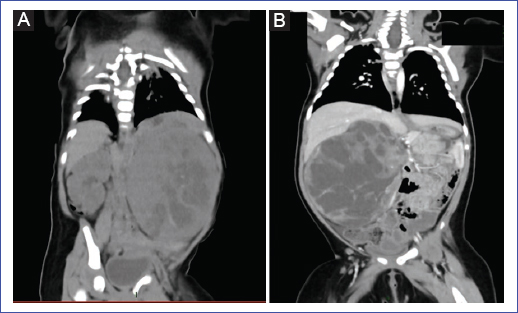
Figura 1. A: tumoración renal izquierda con evidencia de necrosis focal correspondiente al primer caso presentado. B: tumoración renal derecha heterogénea que sobrepasa la línea media en el segundo caso presentado.
El estudio ecográfico abdominal evidenció una extensa lesión sólida en el riñón izquierdo de 86 × 44 × 81 mm de diámetro (163 ml), con múltiples cambios quísticos necróticos en su interior. En la tomografía computarizada abdominal (Fig. 1B) se visualizó una masa sólida en el riñón izquierdo, sin ruptura capsular. Adicionalmente, en el estudio tomográfico de tórax se hallaron lesiones sugestivas de proceso inflamatorio parenquimal incipiente y bula subpleural en el segmento superior del lóbulo derecho. Ganglio en la región axilar derecha de 0,6 × 0,6 cm en sus diámetros mayores, de aspecto inespecífico. Durante su hospitalización presentó episodios de deposiciones con moco y hematuria que remitieron espontáneamente, además de mediciones de presión arterial de 115/80 mmHg, por encima del percentil 95, e hipertensión renal secundaria, tratada con amlodipino.
Se practicó nefrectomía radical izquierda con hallazgos de tumor sólido de 15 × 10 cm aproximadamente, con pedículo vascular con una vena, dos arterias y una arteria polar, un uréter, adenopatías perihiliares y paraaórticos de 1 cm. El resultado de la biopsia detalla NMC de tipo mixto, con invasión del seno renal, sin restos nefrogénicos, invasión linfovascular, vascular y ureteral libre, estadio COG II. Inmunohistoquímica: CD34, ciclina D1, actina, CD99 y P53 negativos, y KI 67 1% (Figs. 2 A–C).
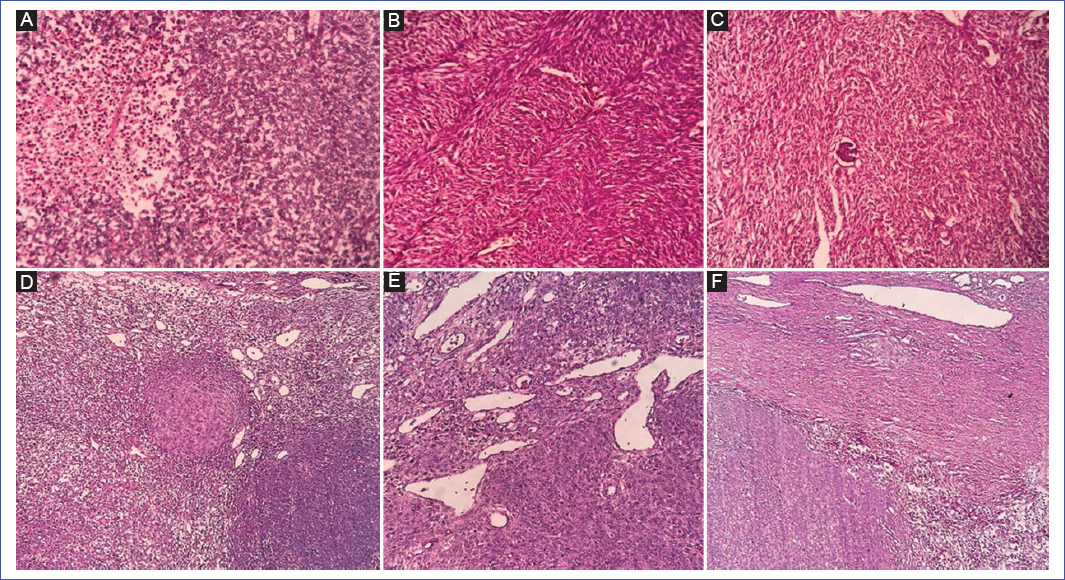
Figura 2. A: alta celularidad con células que presentan núcleos vesiculosos y área de necrosis a la izquierda de la imagen. B: tumor compuesto por numerosos fascículos de células fusiformes. C: glomérulo atrapado en la lesión. D: tejido tumoral con alta celularidad. E: células tumorales con citoplasma escaso, núcleos vesiculosos ovalados y nucléolos poco visibles. F: interfase entre el tumor (parte inferior) y el riñón normal (parte superior).
La paciente permaneció hospitalizada 28 días. El diagnóstico de egreso fue NMC izquierdo e hipertensión arterial secundaria; tratamiento de cefalexina, amlodipino y paracetamol condicional. Durante sus evaluaciones posteriores por nefrología mantiene una función renal conservada y continúa con tratamiento antihipertensivo. Oncología realizó control tomográfico de tórax y abdomen, sin encontrar indicios de tumor. La paciente, actualmente de 5 meses, se encuentra con buena evolución clínica.
Paciente 2
Lactante de sexo femenino, de 6 meses, procedente de Lima. Madre de 29 años sin antecedentes relevantes. Nació por cesárea a término (40 semanas). Acude al hospital por presentar hematuria macroscópica de 10 días de evolución. La ecografía muestra un tumor renal derecho de aspecto mixto, de 99 × 72 mm, en el riñón derecho, con escaso realce al estudio Doppler. Laboratorio: leucocitos 13.600 cel/μl, linfocitos 51%, hemoglobina control postransfusión 15.8 g/dl, urea 13 mg/dl, creatinina 0,34 mg/dl; uroanálisis dentro de parámetros normales. En la tomografía computarizada abdominal completa se visualiza una masa heterogénea en el riñón derecho, de bordes definidos y consistencia mixta, captadora de contraste, de 70 × 61 × 101 mm, que condiciona desplazamiento del espacio perirrenal posterior. Inicialmente se sospechó tumor de Wilms.
La paciente fue sometida a nefrectomía radical derecha, encontrándose una tumoración de 12 × 8 cm, adherida a la cara inferior del hígado. El resultado histopatológico concluyó NMC de tipo celular, confinado al parénquima renal, sin invasión linfovascular y con ganglios linfáticos libres de neoplasia (Figs. 2 D–F). El tiempo de hospitalización fue de 43 días, siendo su evolución favorable. En controles posteriores no presentó complicaciones y tuvo buena respuesta al seguimiento. Fue evaluada por cardiología por antecedente de taquicardia poscirugía, la cual cedió en los siguientes controles.
Discusión
El NMC es el tumor renal más común en neonatos y lactantes, representando el 75% de los casos en los primeros 3 meses de vida, a diferencia del tumor de Wilms, que es más frecuente entre 1 y 4 años6. Nuestros dos casos fueron lactantes menores de 6 meses que presentaron masa abdominal y hematuria franca.
La histopatología del NMC está bien caracterizada. El subtipo celular muestra mayor celularidad, alto índice mitótico y mayor invasión, con áreas de fibrosis, hemorragia y necrosis, mientras que el tipo clásico es más circunscrito, con células fibroblásticas, depósitos de colágeno y baja actividad mitótica7.
La mayoría de los casos de NMC se manejan solo con cirugía, con una tasa de supervivencia promedio del 98%1, tal como sucedió en nuestros dos casos reportados. La nefrectomía radical es el procedimiento de elección y requiere una disección cuidadosa para evitar la ruptura tumoral, lo que podría complicar el cuadro8. Por ello, algunos autores desaconsejan la biopsia guiada por imagen2. Asimismo, se debe eliminar la grasa perirrenal para evitar la presencia de neoplasia residual.
Se deben tener en cuenta los riesgos quirúrgicos producto de la nefrectomía radical en pacientes de temprana edad, ya que se han reportado complicaciones quirúrgicas en el 23% de los casos de NMC2. Además, hasta el 60% de los pacientes con tumores renales congénitos pueden desarrollar hipertensión secundaria a hiperreninemia9. En este estudio, una paciente presentó hipertensión arterial y la otra tuvo taquicardia posquirúrgica, ambas con evolución favorable. Si bien ambos casos tuvieron una buena evolución cardiológica, es importante alertar al equipo a cargo de la operación sobre este tipo de consideraciones y potenciales complicaciones cardiovasculares.
Aunque raros, se han publicado casos de recurrencia y metástasis a distancia en subtipos histológicos celulares de NMC, principalmente en aquellos con márgenes quirúrgicos positivos, rotura tumoral intraoperatoria o diagnóstico tardío10. Solo si se presenta recurrencia se sugiere optar por el uso de quimioterapia adyuvante. Debido al riesgo, aunque mínimo, de recurrencia, se recomienda un seguimiento clínico y ecográfico estrecho, preferentemente cada 3 meses en los primeros 12 meses después de la cirugía primaria, y cada 6 meses hasta los 24 meses10.
Conclusiones
El diagnóstico diferencial de tumores renales en pediatría incluye NMC, tumor de Wilms, sarcoma de células claras y tumor rabdoide, entre otros. La rareza del NMC y su similitud con otros tumores dificultan el diagnóstico, por lo que debe considerarse ante la presencia de una masa renal en neonatos. Es un tumor de bajo grado de malignidad, con excelente pronóstico tras la nefrectomía radical. El enfoque quirúrgico debe minimizar las recurrencias y garantizar un seguimiento estrecho, en especial en las variantes celulares y mixtas.
Financiamiento
Los autores declaran no haber recibido financiamiento para este estudio.
Conflicto de intereses
Los autores declaran no tener conflicto de intereses.
Consideraciones éticas
Protección de personas y animales. Los autores declaran que los procedimientos seguidos se conformaron a las normas éticas del comité de experimentación humana responsable y de acuerdo con la Asociación Médica Mundial y la Declaración de Helsinki. Los procedimientos fueron autorizados por el Comité de Ética de la institución.
Confidencialidad, consentimiento informado y aprobación ética. Los autores han seguido los protocolos de confidencialidad de su institución, han obtenido el consentimiento informado de los pacientes, y cuentan con la aprobación del Comité de Ética. Se han seguido las recomendaciones de las guías SAGER, según la naturaleza del estudio. El estudio se someterá al Comité de Ética en Investigación del Instituto Nacional de Salud del Niño.
Declaración sobre el uso de inteligencia artificial. Los autores declaran que no utilizaron ningún tipo de inteligencia artificial generativa para la redacción de este manuscrito.
